
DNAの抽出
DNA抽出サンプルの準備
- 藻体の一部(1cm角)をちぎり、滅菌海水入りディスポシャーレに入れ,実体顕微鏡で確認しながら筆で汚れを落とす。(汚れがひどければ繰り返す)
- オートクレーブ済みのエッペンチューブ(名前を忘れずに)にクリーニングした藻体を入れ,キムワイプで水気を取る。
- 使ったピンセットや筆は70%エタノールにつけて,キムワイプで拭いてケースに戻す。
- デシケーターに入れ真空ポンプで空気を抜きながらドライアップ(10分)。
- 証拠標本として押し葉標本を作成する。
DNA抽出(QIAGEN DNeasy Plant Mini Kit)
- エッペンチューブ(エッペン)にペッスルをさす。
- 液体窒素をコップに入れる。サンプルの入ったエッペンをトレーに立て,液体窒素をゆっくり注ぎ,軽く沸騰したら,再度いっぱい入れる。液体窒素が半分ぐらいまで蒸発したら,ペッスルをいれ,冷やす。液体窒素が無くなる寸前で,ペッスルをグリグリしてサンプルを磨りつぶす(ペッスルは出さない)。
- 破砕したサンプルにAP1を300μlいれ,ペッスルを回しながら抜き,フタをしっかりして,ボルテックス。
- 65℃で10分インキュベート。2-3分毎にチューブを軽く撹拌。各種チューブ用意。 (この時AEをsample数×100μlをエッペンに取り同じく65℃)
- 軽くチビタン。ゆっくりフタを開けて,P3 (AP2)を130μl加え、ボルテックス、氷上5分、 14000rpm遠心5分
- 上澄み300μlをQIAカラム(紫色)に移し、14000rpm遠心2分
- 450μlのAW1(AP3/E)を新しいエッペンに入れておく。青ピペットを850μにし,紫カラムを捨て,落液を全量吸い,AP3/E入りのエッペンに移し,やさしくピペッティング5回,白カラムに全量移す。10000rpm遠心1分
- 落液・collection tubeを捨てて,カラムだけ新しいcollection tubeに移し、カラムにAW2(AW)を500μl入れる。10000rpm遠心1分。
- 落液を捨て、同じcollection tubeを再装着し,カラムにAWを500μl入れる。14000rpm遠心2分。
- 静かにチューブを机に持ってきて,落液・collection tubeを捨てて,カラムを新しいエッペンに移し、65℃に熱したAEを80μl膜にかける。
- 5分室温で待ち10000rpm遠心1分
関東化学シカジーニアスDNA抽出試薬
(短時間で、容易で、安価、長期保存にはむかない)
- 上述のように液体窒素で藻体をつぶす。
- 1サンプル試薬a液10μl, 試薬b液100μlの比率で混合液を作り、すりつぶした藻体に110μlずつ入れる。軽く指で弾いて混合。
- 72℃ 20min, 94℃ 3mインキュベート
- 14000rpm 3min遠心し、上澄み80μlを別エッペンに。
PCR反応
Takara Ex Taq
- 氷を準備し、PCRトレイをのせておく。
- 0.2mlPCR用チューブに名前を書く。
- 以下の鋳型DNA以外をすべて混合しPCR用チュ-ブに29 μl分注する(ネガティブコントロールも)。
10xPCR buffer 30.0μl
dNTPmix 24.0μl
DMSO *1 15.0μl
DW 220.0μl
Primer No.1 *2 5.0μl
Primer No.2 *2 5.0μl
Taq polymerase 1.5μl
*1 18SrDNA, nrITSのみ入れ、他はこの分DW
*2 プライマー20pmol/μl (20 μM)
4. 抽出DNAを1~2 μl加えボル。
5. サーマルサイクラ-各種プログラムで増幅。
94℃ 1分 (この間にサンプルをセット)
94℃ 15秒、 50℃ 15秒、 68℃ 30秒 *3、 ×45サイクル
4℃ 保持
*3 extentionの目安は1kb=1imin
→基本プログラムで増幅しない場合は要相談
PCR産物の電気泳動
1%アガロースゲルの作製(短い断片同士の距離を出したい場合は濃度をあげる)
- ゲルトレイスタンドにゲルトレイをはめて、コームを2本挿す。2本目は下から4本目の細線の所。(16サンプル以下→小さいゲルトレイ)
- アガロース150 mg、1xTAE Buffer 15 ml(10xTAEじゃない)をはかりとり、三角フラスコに入れて90秒チン。 (大ゲルトレイの場合:アガロース250 mg、TAE Buffer 25 ml。1分ほどで沸騰するので取り出して軽く振り沸騰を鎮め、残り時間チン)
- 良くかき混ぜ、しずかにトレイに流し入れコームをさす。泡がないか確認。
- 30分待ってからTAE を少し入れてコームをはずす。
電気泳動
- パラフィルムにLoadingBuffer 1μづつ分滴。
- 必要なだけゲルをカット。使わないゲルは三角フラスコへ。
- PCR産物 4μをとりLoadingBufferと混ぜてLoading。
- Mupid-2 plus を使用。135V 10分 電気泳動。(感電に注意)
- ゲルを取り出しエチブロに10分。(ディスポグローブを必ず着用)
- サランラップにゲルを載せてトランスイルミネーターで観察。
- ポラで写真を撮る。ゲルはシンクの可燃ゴミ。
Takara Mighty Amp
増幅が困難な場合、試す。
2xBuffer 25.0 μl
Primer 1 (20μM) 0.5 μl
Primer 2 (20μM) 0.5 μl
Mighty Amp DNA polymerase 1.0 μl
temDNA 1.0 μl
DW 22.0 μl
Total 50.0 μl
PCRプログラム
98℃ 2分 (Hot Start)
98℃ 10秒、 60℃ 15秒、 68℃ 1 min/1kb、 ×40 サイクル
4℃で保持
PCR産物の精製
Wizard® SV Gel and PCR Clean-Up System – Promega
[ゲルの切り出しの場合]
- 切り出したゲルを5mlエッペンに入れ,ゲル10mgあたり10μlのMenbrane Binding Solutionを入れる。55℃8分。(溶ければOK)
それ以降は下の2)以降と同じ。
[PCR産物の場合]
- PCR反応液と同量(26μl)のMenbraneBindingSolutionをPCR産物の入ったPCRtubeに入れ,ボル・チビ。
- SV minicolumnをCollection Tubeに入れ,sample番号をどうにか書いて、1の全量をカラムに入れ,室温1分。
- 遠心13,500rpm,1分。
- 700μlのMembrane Wash Solution(エタノールが加えられたもの)を入れ遠心13,500rpm,1分。落液を捨て,再度Collection Tubeに装着。
- 500μlのMembrane Wash Solution(エタノールが加えられたもの)を入れ遠心14,000rpm,5分。
- 落液を捨て,再装着し、からのまま遠心13,500rpm,1分。
- やさしく,SV minicolumnを新しい1.5mlエッペンに装着し,Nuclease-free Waterを50μ入れ,室温1分。13,500rpm,1分。
Cycle Sequence、精製
BigDye v.3.1 34.0µl (分注済み1本分)
DW 26.0µl
5x Buffer 17.0µl
- 上記ミックスをボルテックス、チビタン、4.5 µlずつPCRチューブに分注(残りはフタを黒く塗って箱に戻す)
- primer(1-2picoM) 1.6 µl (1サンプルF or Rどっちかのみ!)
- tempDNA 3.9 µl
を加え、ボルテックス、チビタン PCRマシーンで「Cycle Seq」約2.5時間
[精製]
12sample分
DW 130 µl
EDTA(125mM) 26 µl
酢酸Na(3M) 26 µl
- ボル、14µ づつPCRチューブに分注してボル、エタノール50 µl 加えボル
- 室温で遮光 15min待ち、12500rpm、20min、5℃
- 上澄みを捨てて、70%エタノールを100µ、12500rpm、5min、5℃
- 上澄みを捨てて、アルミ箔で覆い、Dry up 10min
0.5M EDTA (pH 8.0) (冷蔵)
EDTA=ethlenediaminetetraacetic acid・2Na・2H2O; MW=372.24
EDTA 46.5 g
DW 200 ml +α(メスアップ)
計 250 ml
- ビーカーに水を入れスタラーで撹拌しながらEDTAを加える。
- pHメーターで測りながら粒上のNaOHを1つぶずつ加え、EDTAを溶かす。(最終的に4,5つぶ)
- pH8に近づいてきたら5N NaOHを加え、pHを調整する。
- メスアップ。試薬ビンに移し、オートクレーブ。
125mM EDTA (もとは冷蔵、小分け常温可)
- 0.5M EDTAを純水で4倍に希釈
- オートクレーブ。
3M 酢酸ナトリウム (pH 5.2, pH 7.0) (もとは冷蔵、小分け常温可)
(Cycle sequence精製時はpH 5.2) sodium acetate (NaOac)・3H2O; MW=136.08
NaOAc・3H2O 3M 20.4 g
DW 40 ml +α
計 250 ml
氷酢酸(酢酸)(pH調整用) pH 7.0の時 約64μl
pH 5.2の時 約5.7ml
- ビーカーに水を入れ、スタラ-で撹拌しながらNaOAc・3H2Oを溶かす。
- 目的のpHに合わせる。
- メスアップ。試薬ビンに移し、オートクレーブ。
サブクロ2007
LBA培地の作成 (10 plates用)
- LB agar 8gをDW250mlにいれ、そのままオートクレーブ(121℃15分)
- 55℃までに12.5mg/mlに調整されたアンピシリンを1ml入れ混ぜる.
- プレートに適量注ぎゲル化させる。
- アンピシリンは失活しやすいので2週間が限度
Cloning samplesの用意
通常のPCR sampleを用意。Final Extension (72℃10分とか)があるほうが良い。
ライゲーション反応
2x Rapid ligation buffer 5.0 μl
pGEM-T Easy vector 0.5 μl
PCR産物 4.0 μl
T4 DNA Ligase 1.0 μl
上記反応液を室温で1時間反応させる。
形質転換
- セルを-80℃から取り出して氷中で解凍。
- ライゲーション反応液をエッペンチューブに分注。
- セルを2のエッペンチューブに50μlずつ分注し、強く指で3回はじく。
- 氷上30分。
- 2℃で45~50秒間ヒートショック。
- 氷上2分。
- SOC(冷凍庫の別袋 黄色の液体1.5チューブ)を250μlずついれ、37℃で1.5~2時間。
- (X-galをLB plateに60μlづつ塗り乾燥)
- 120μlずつ、プレートに撒く。
- 37℃、16-24時間培養。
Colony Direct PCR
LBplate上の白いconolyを白チップでちょんっとつけて
通常のPCRsamplにつっこみピペッティング。 [30cycle]
Grow in a liquid LB medium
乾熱した試験管にLB培地を3mLディスポピペットで入れる。
LBplate上の白いconolyをプラスチック白金耳でちょんっとつけて試験管につける。
37℃、一晩 (各器具はブリーチ)
QIAprep Spin Miniprep Kit Protocol
(Kitが届いたら)
- Rnasae AをBuffer P1に入れ混ぜ、2-8℃で保存。Buffer PEにエタノールを24mL。(注意事項)P2は強く振らない、すぐ蓋を閉める。P2, N3, PBは手袋着用。
- 遠心は13000rpm、室温
- 試験管を回し1.5mLをtubeにいれ遠心1分、上澄みをブリーチ専用ディスポにいれる。
- 残りを入れて(1)をもう一度。ペレット状cellに250μlのP1でピペットで再濁
- 250μlのP2を入れ5回やさしくtubeをひっくり返す
- 350μlのN3を入れ5回やさしくtubeをひっくり返す
- 10分遠心し、上澄みをQIAprep Spin Columへ
- 1分遠心、flow-throughは捨てる
- 750μlのPEを入れ、1分遠心
- flow-throughは捨て、空のまま1分遠心
- 新しいtubeにサンプル名を書き、 Columを装着
- 50μlのEBを入れ、1分待って、1分遠心
シークエンサーの使い方
予約
6階のシークエンサー室前のカレンダーで予約を入れる。 ラン回数と名前を書き使用時間分の線を引く。
サンプル準備
- 分注済みのHi-Di Formamideを-20℃から必要量出して溶かす。
- 乾燥済みのサイクルシークエンスサンプルにとけたHi-Di Formamideを20μLいれ、ボル・チビ。
- 96well プレートに全量移す。1A, 1B, 1C…..2A, 2Bの順。 16サンプルずつキャピラリーで吸われるので、残穴にもHi-Di Formamideを20μLいれる。プレートセプタをしっかりはめる。
パソコンでの準備
- パソコンの電源、シークエンサーの電源の順で入れる。Run 3130 Data Collection をダブルクリック。
- 左アイコン左の+アイコンを押してすべて表示させる。 Plate Managerを押して、自分のPlate IDをダブルクリック。 (追加する場合は、自分のPlate IDをクリック(青変)し、下の「Duplication」を押して、ID名を変更して、表を全て選んで「Edit」から「Clear row」で白紙にもどし、「OK」)
- 表の「sample name」にサンプル名、「results Group1」にオーナー名、「Instrument Protocol 1」には、600bp以下の場合は「UltraSeq36_POP7_v3」、それ以上の場合は「RapidSeq36_POP7_v3」、 [Analysis Protocol 1]には「3130KB_POP7_v3」を選択し、「OK」。(コピペ可能)
シークエンサーでの準備
- プレートを黒いプレートベースにはめて、白いプレートリテイナーを上から「カチッ」とはめる。
- シークエンサー正面左下「TRAY」ボタンを押すとオートサンプラーが手前に出るので、ドアを開けてプレートベースの凹を奥にして取り付ける。
- ドアを閉めれば、オートサンプラーは自動的に元の位置に戻る。
ポリマーが足りなそうだったら、冷蔵補から「POP7」をだして、気泡をださないように静かに充填する。 2週間に1回は、オートサンプラーの水・Bufferおよび左下の陽極バッファーリザーバのBufferを交換する。オートサンプラーの左手前が Bufferで残りの3つがDW用。Bufferはx10を冷蔵庫から取り出し、50mLコニカル(専用がシークエンサー室にあり)にx10 Bufferを3.25mlをピペットでとり、32.5mlまでDWでメスアップ。陽極バッファーリザーバに先に入れ、残りを陰極へ。セプタはしっかり。
注意!シークエンサー内の水漏れは漏電の原因となり危険です。しっかりこぼれた水気は取り除きましょう。
月はじめの人は、ウォーターシール部分の水を充填。専用注射器にDWを入れ、シリンジフィッティングに注射器をねじつけて廃液フィッティングをゆるめてキムワイプでおさえながらDWを充填し、廃液フィッティングを閉め、注茶器を取り外す。
ラン
- [Run Scheduler]で打ち込んだPlate Nameを選びクリック(青変)。
- 右の図を押し、マップが黄から緑へ変わったら、左上([File]の下)の△が緑に変わるので、押すとランが開始。
- データは自動的に解析され、保存される。必要に応じてプリントアウト、メモリに落とし、マックへ その日のうちに使用者がいない場合は、シークエンサー、パソコンの順に電源を落とす。 利用者ノートに利用記録を記入する。
初めての人は「ユーザーガイド」を熟読し経験者に見てもらいながら行うこと。 キャピラリー交換や不明な点は、自分の判断で行わず、必ず教員の指示を仰ぐこと。
アライメント
- パソコンを512から持ってくる(テーブルで1台)
- パソコンを立ち上げて、フォルダをコピー(2012-07-11_Shimada-lab142
- BioEditでfas(アライメントファイル)を開く
- Sequences in colorを押す
- FileからOpen B03_名前_2012-07-11.ab1
- 波形を確認 (汚い部分は後で修正する必要あり)
- 下に開いたファイルをクリック 番号をクリック EditからCopy sequence
- アライメントファイルをクリック EditからPaste sequence
- ModeをEditに 右隣はInsert
- 自分の配列の最初の部分をdeleteあわせる 順次ハイフンを入れてあわせる
- あわせながら近い配列に移動させる(数字をクリック左押しながらスライド)
- 後ろのいらない部分もdeleteであわせ、保存
系統樹の構築
- MEGA5をダブルクリック
- FileからOpen A File/Sesson ITS2Ulva2012をダブルクリック
- Analyzeをクリック OKをクリック Noをクリック
- PhylogenyからConstruct/Test Neighbor-Joining Treeをクリック
- Yesをクリック
- Gaps/Missing Data Treatment はPairwise deletionを選択(黄色部分をクリック、選択)
- Computeをクリック
- DNA鑑定の結果を確認 ImageからCopy to Clipboard
- スタートからパワポを開き、デザインからスライドの向きを縦に
- いらないのwindowは消して、ペースト
- 上下をファイルサイズにあわせて 右クリック グループ解除 2回
- 加工して終了 プリントアウトして提出
RNAの抽出
- 生100mg藻体入りのチューブに液体窒素をゆっくり適量入れ、ドリルで破砕 (液体窒素使用時は換気に注意!)
- 急いで破砕サンプルに450μづつA液を分注、ボルテックス
- 全量をQIAshredderカラム(紫色)に移し、最速遠心2分
- 下のゴミを採らないように注意して上澄み450μlを新しいエッペンに移し、 0.5倍(225μl)のEthanolを加え、急いでピペッティング
- 急いで全量をRneasyカラム(桃色)に移し、静かにふたを閉め、 10000rpm遠心15秒、落射液を捨て、同じcollection tubeを再装着
- カラムにBuffer RW1を700μl入れ、静かにふたを閉め、10000rpm遠心15秒 (落射液を捨て、collection tubeも捨てる)
- 新しいcollection tubeを付け、カラムにBuffer RPEを500μl入れ、静かにふたを閉め、 10000rpm遠心15秒 (落射液を捨て、同じcollection tubeを再装着)
- カラムにBuffer RPEを500μl入れ、静かにふたを閉め、10000rpm遠心2分 (溶出液・collection tubeを捨てる)
- カラムに新しいエッペン(番号記入)を付け、DW30μlを膜にかける、 静かにふたを閉め、10000rpm遠心1分 (必要なら9を繰り返す)
- Nano Dropで濃度を測定
- 3M Naアセテイト(1/10倍量)+エタノール(2.5倍量)で-80度にストック
mRNA FingerPrinting Kit
- 3‘-Anchor primer毎に下記の溶液を準備する。 DEPC Water 8.0μl 3’-Anchor Primer 1.0μl Total RNA 1.0μl (2.5μg)
- 70℃, 10min熱変性。
- 氷冷3min。スピンダウン。
- 下記の溶液を加える。 DEPC Water 2.0μl 10×RT Buffer 2.0μl 25mM MgCl2 2.0μl 100mM DTT 2.0μl 10mM dNTP 1.0μl Reverse Transcriptase 1.0μl
- サーマルサイクラーで『cDNA』を実行。
- PCR Purification Kitで精製。最終の液量は45μlとし、ここに3’-Anchor Primerを5μl加える。
- 『D.D. METHOD』のPCRを実行する。 10% ポリアクリルアミドゲル (大ゲル一枚分の分量) 30% ポリアクリルアミド 8.340ml 1.5M Tris-HCl (pH 8.9) 6.260ml DW 10.185ml 10% APS 200μl TEMED 15μl Total 25ml
- 差が出たバンドを切り出して、アクリルアミドからDNAを抽出。
- *『ポリアクリルアミドゲルからのDNA断片の回収』を参照
- Program『2nd D.D.』を用いて再度PCR。
- Anchor Primerは0.2μl、Arbitrary Primerは0.5μl、DNAは1μl使用する。
- ゲルを切り出し、クローニング。
ポリアクリルアミドゲルからのDNA断片の回収
- 目的のゲルをカッターで切り出す。
- 切り出したゲルを-20℃で凍らせる。
- 凍ったゲルに液体窒素を加え、ペッスルで破砕。
- 600μlのBufferを加えてVortexし、ウォーターバスで37℃、1晩インキュベート。 (Buffer=0.5M酢酸アンモニウム1mMEDTA 遺伝子冷蔵庫070801)
- 14000rpm、5min. 上澄みを新しいチューブに移す。
- ペレットにさらに150μlのBufferを加えVortex。
- 14000rpm、5min. 上澄みを5の上澄みと混ぜる。
- 上澄みを300μlずつチューブに移し、ここに30μlの3M 酢酸ナトリウム、750μlの100%エタノールを加えて、室温で15min.
- 4℃, 14000rpm, 20min.
- 上澄みを捨て70%エタノールを1ml加える。
- 4℃, 14000rpm, 5min.
- 乾燥させ、20μlのDEPC WATER or EB などに溶かす。
3′ RACE法
1st strand cDNAの合成
- RNAサンプル/プライマーの混合液を作る。
- Total RNA(1ng~5μg) dNTP mix(10mM) 1μl 3’-RACE CDS Primer 1μl DEPC処理水 Total 12μl
- RNAサンプル/プライマー混合液を65℃で5分間インキュベーとする。
- 氷上に移して1分以上置く。
- 以下の試薬を順に混合し、サンプル分のMixを作る。
- RNAサンプル/プライマー混合液 12μl 5×cDNA buffer 4μl DTT(0.1Mストック) 2μl Total 18μl
- 42℃で2~5分間プレインキュベートする。
- 逆転写酵素を1μl加え、チューブの内容物をピペッティングでよく混合する。
- 42℃で50分間インキュベート。
- 70℃で15分間インキュベートし、反応を止める。
- 氷上で冷やした後、軽く遠心して反応液をチューブのそこに集める。
- そのままPCRに用いるか、-20℃でストックする。
3’RACE法
- 通常のPCRmix(primer抜き)を作成する(DMSOの有無はケースバイケース)。
- 1.で作ったMixをdegenerate primer・3’adapter primerが1μlずつ入ったPCRチューブに分注する。
- 最初の熱変性だけ2分長く取り、後は通常のPCRを行う([1PCR]のprogramを用いればよい)。 アニーリング温度は個々のプライマーによって決定する。サイクルは25~30回程度。
- アガロースゲルによる電気泳動でバンドを確認する。
- バンドが確認できたらサブクロして、PCRを行う。
- PCR産物を精製しCycle Sequenceを行い(この時のprimerはプラスミドのものを使ったほうが増幅は安定する)、シーケンサーへ。
- シーケンスにpoly(A)が確認できたらmRNAから増幅できたものだと考えてよい。配列から5’RACE用のprimerを作成する。
5′ RACE法
cDNAの合成
GSP1 (3’で決めた配列から作製するprimer) 2.5pmoles Sample RNA 1-5μg DEPC-treated water 15.5μl
- このmixを70℃、10min
- component (μl) 10×PCR buffer 2.5 25mM MgCl2 2.5 10mM dNTP mix 1 0.1 M DTT 2.5 Final volume 8.5 これを温めたsampleに加え、42℃、1min
- SuperScriptⅡを1μl加えて『cDNA』のプログラム
S.N.A.P Column Purification of cDNA
- binding solutionを室温に戻す
- サンプル数×50μlのsterilized, distilled waterを65℃に温めておく。
- 120μlのbinding solutionをfirst strand sampleに加える。
- sampleをS.N.A.P columnに移し、13000rpmで20sec。
- カラムをはずし、落液はcDNAの存在が確認されるまでPCRチューブに保存。
- カラムに4℃のwash bufferを400μl加え、13000rpm、20sec、落液を捨てる(×3)。
- 400μl、4℃の70%エタノールを加えて、13000rpm、20sec、落液を捨てる(×2)。
- 空のまま13000rpm、1分。
- カラムに新しいチューブをつけて、65℃に温めたsterilized, distilled waterを50μl加え、13000rpm、20sec。
TdT Tailing of cDNA
- 以下の溶液をPCRチューブに入れてやさしく混合する。 DEPC-trerted water 6.5μl 5× tailing buffer 5μl 2mM dCTP 2.5μl cDNA sample 10μl Final volume 24μl
- 2~3分94℃でインキュベート。1分間氷上。チビタン、氷上。
- TdTを1μl加え、37℃で10分間。
- GSP2(GSP1より5’で作製したprimer)とキットのアダプターprimerでPCR、シークエンス アライメントで3’とかぶるか確認。
Northern Blot法
ECL Direct Nucleic Acid Labeling and Detection System プローブの標識
- 標識するDNAをキット附属の水(またはDEPC WATER)で10ng/μlに希釈する。
- 100ngのDNA(RNA)サンプル(10μl)をとり、沸騰水中で5min, 熱変性。
- サンプルを氷中で5min急冷後、スピンダウン。
- 熱変性したサンプル10μlのDNA labeling reagentを加え、穏やかに完全に混和。
- Labeling reagentと等量(10μl)のglutaraldehyde solutionを加え、完全に混和、スピンダウン。
- 37℃、10minインキュベート。
- 作成したプローブは直ちに使用するか、氷中に保存し、10~15min以内に使用する。
ハイブリダイゼーション 1次洗浄バッファー Urea 36g SDS 0.4g
| 20×SSC 2.5ml |
Fill up to 100ml (DW) 2次洗浄バッファー
| 20×SSC 10ml |
Fill up to 100ml (DW)
- ハイブリダイゼーションバッファーを室温で必要量はかり、最終濃度5%となるようにBlocking reagentを、0.5MになるようにNaClを添加し42℃に温めながら攪拌し、完全に溶解させる。(バッファーの量はメンブレンの面積×0.125ml程度)
- 42℃のハイブリダイゼーションバッファーの中にメンブレンをいれ、1hrプレハイブリダイゼーション。
- 標識プローブを添加。穏やかに振盪しながら42℃で一晩ハイブリダイゼーション。(ブロットに直接かからないようにバッファーの一部を取り出してプローブと混和して戻すと良い)
- 一次洗浄バッファーを42℃に温めておき、ブロットを浸し42℃を越えないように20min,振盪。(バッファー量はメンブレンの面積×2~5ml)
- 4を繰り返す。
- きれいな容器にメンブレンを移し2次洗浄バッファーを入れ室温で5min, 振盪。
- 6を繰り返す。
- 洗浄後はサランラップに包んでメンブレンが乾かないように2~8℃で保存。
シグナルの検出
- Detection reagent1とDetection reagent2を等量混合し、メンブレン上の余計な2次洗浄バッファーを除き、DNAがブロットされている面に検出試薬を添加する。(検出試薬の量はメンブレンの面積×0.125ml程度)
- 室温で1min。
- メンブレンの角を持って余計な検出試薬を除き、気泡が入らないようにサランラップで包む。
- ブロットが上になるようにサンプルをフィルムカセットの中におきHyper film ECLをセットする(暗中)。
- 1min, 露光させる。
- 現像、定着液5min、洗浄30min。
Northern Blot法
試薬作成時には専用の器具を用いること。 準備 ~試薬調整~
- 20×MOPS (pH 7.0) CH3COONa・3H2O 1.36g MOPS 8.37g
| EDTA・2Na・2H2O 0.75g |
- Fill up to 100ml(DW)
- 2.20×SSC (pH 7.0)
- NaCl 87.6g
| Sodium Citrate 50g |
- Fill up to 500ml(DW)
- 作成後オートクレイブ処理したほうがよい。
☆ゲルの作成 1% 100ml
- アガロース 1.0g 20×MOPS 5ml DW 50ml を混合し、レンジで温めて溶かし、さらに残りのDW27mlを加えて温める。
- 55℃程度まで冷まし、ホルムアルデヒド18mlを加える。
- 型に流す。
注・ホルムアルデヒドは揮発しやすく有害!ゲルはドラフト内で固める。
☆Sample調整
Sample Buffer 20sample分
0.4mg/ml EtBr 20μl
20×MOPS 10μl
ホルムアミド 200μl
ホルムアルデヒド 70μl
- S.B.15μlとRNA 5μlを混合し、65℃、15minで変性させる。
- ローディングバッファーと混合し、アプライ。
☆電気泳動
- 1×MOPSで泳動槽を満たし、電気泳動 (60Vで2hr程度) 。
- 終了後ゲルをDWで軽く洗い、さらに20×SSCに5min浸す×2回(この項は省略可)。
☆ブロッティング
- Hybond H+をゲルの大きさに切る。
- 下図のようにセットする。
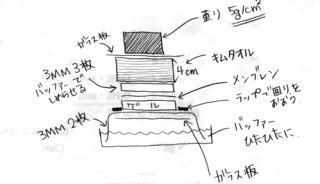 注・ゲルとメンブレンの間に気泡を入れない。 ウェルを下側にする。
注・ゲルとメンブレンの間に気泡を入れない。 ウェルを下側にする。
☆プローブの作成
Amersham社Gene Images Random Prime Labeling Kitを使用した場合
- 濃度2-25ng/μlのDNA溶液を用意する。
- DNA溶液を5min、シャンメルし氷冷。
- Nucleotide mix 10μl Primer 5μl Denatured DNA 50ng Klenow 1μl Fill up to 50μl (DEPC Water)
- ピペッティングでやさしく混合。
- 37℃、1hrで反応させる。
☆プローブの精製